利用Cas9 RNP技术制备B2M和PD-1高效率双基因敲除的人原代T淋巴细胞
【摘要】目的 观察Cas9 RNP技术制备B2M和PD-1双基因敲除的人原代T淋巴细胞(以下简称T细胞)的效率。方法 设计单向导RNA(sgRNA)后克隆至载体质粒,经转染HEK293T细胞后筛选出高效率的sgRNA;先后构建B2M及PD-1单基因敲除及同时敲除双基因的T细胞,分别通过Sanger测序、TA克隆及流式细胞术等方法验证编辑效率。结果 成功构建包含sgRNA的载体质粒并经初筛获得高效率的B2M sgRNA1及PD-1 sgRNA1;在T细胞中验证B2M和PD-1单基因敲除效率均高达90%;构建双基因敲除T细胞后经验证基因层面的编辑效率高达90%,B2M及PD-1蛋白的表达下调率分别达86%、89%。结论 利用Cas9 RNP技术可制备B2M和PD-1高效率双基因敲除的T细胞。
【关键词】Cas9;核糖核蛋白;PD-1;&β;2-微球蛋白;T淋巴细胞
0
引言
近年来肿瘤免疫治疗的飞速发展改变了肿瘤治疗的格局,其中嵌合抗原受体(chimeric antigen receptor, CAR)T细胞在临床上已经取得了显著的成效,两款自体CAR T细胞产品已获美国食品药品监督管理局(Food and Drug Administration, FDA)批准上市。但是,因为肿瘤免疫抑制性微环境的存在,大部分患者无法提供足量且高质量的T细胞进行治疗,所以这种直接取材于患者自身或造血干细胞移植受体中的干细胞供体的高度个性化的策略限制了其更广泛的应用[1-2]。此外,制备自体T细胞产品所需的长生产周期和高昂费用,也是患者所面临的实际问题。虽然使用来自健康供体经过遗传修饰的同种异体T细胞作为现成的“通用”T细胞能快速治疗多个患者,理论上可以规避这些限制,但输注同种异体T细胞所引起的移植排斥反应又会引发新的临床安全等问题。
人类白细胞抗原(human leukocyte antigen,HLA)分子包括Ⅰ类(HLA-Ⅰ)和Ⅱ类(HLA-Ⅱ)抗原。其中,HLA-Ⅰ类分子负责将抗原肽呈递给CD8+T细胞,被激活的CD8+T细胞可以攻击异己细胞,介导免疫排斥反应[3]。由&β;2-微球蛋白(β 2-microglobulin, B2M)基因编码的B2M蛋白构成HLA-Ⅰ类分子的轻链。B2M基因的敲除可以导致HLA-Ⅰ类分子功能丧失,显著降低了B2M缺失人类胚胎干细胞(human embryonic stem cells,hESCs)对CD8+T细胞的免疫原性[4]。程序性细胞死亡蛋白-1(programmed cell death protein 1,PD-1)基因可表达于T细胞表面,通过与程序性细胞死亡配体1(programmed cell death ligand 1, PD-L1)结合而抑制T细胞的活化及细胞因子的产生,在维持机体的外周耐受上发挥至关重要的作用,而肿瘤细胞利用此机制形成免疫逃逸。PD-1/PD-L1抑制剂正是通过阻断此信号通路重新激活T细胞,从而达到抗肿瘤效果。Rupp等[5]发现肿瘤细胞PD-L1表达可使人CAR T细胞功能低下,而通过基因编辑技术敲除CAR T细胞及T细胞受体(T cell receptor, TCR)T细胞的PD-1基因可显著增强其体外肿瘤细胞杀伤功能[6]。此外,临床前研究已经证实特异性阻断PD-1分子的抑制剂可以有效地增强CAR T细胞的治疗效果[7]。
本研究利用成簇规律间隔短回文重复序列相关蛋白9(clustered regularly interspaced short palindromic repeats-associated protein 9,CRISPR Cas9)核糖核蛋白(ribonucleoprotein, RNP)技术制备B2M和PD-1双基因敲除的人原代T细胞,以消除HLA-Ⅰ类分子所介导的同种异体移植免疫排斥反应,为减轻甚至消除“通用”CAR T细胞的免疫排斥反应,并同时增强T细胞的功能提供理论基础。
1
材料与方法
1.1 材料与试剂
HEK293T细胞为本实验室所有;人原代T淋巴细胞(以下简称T细胞)分离自健康供体血液;pGL3-U6-PGK-puromycin及spCas9质粒来自上海科技大学黄行许教授实验室。单向导RNA(single-guide RNA, sgRNA)及Cas9蛋白由南京金斯瑞生物科技公司化学合成,引物由苏州金唯智生物科技有限公司合成。感受态大肠杆菌DH5&α;菌株、DM2000 DNA marker购自北京康为世纪有限公司;高保真限制性内切酶BsaⅠ-HF(#B7002S)、CutsMart Buffer、NEBuffer 2购自美国New England Biolabs公司;Phanta Max Super-Fidelity DNA Polymerase PCR试剂框购自南京诺唯赞生物科技有限公司;DNA A-Tailing Kit、pMDTM19-T Vector Cloning Kit、DNA Ligation Kit Ver 2.1、6×Loading Buffer购自TaKaRa公司;2×Hieff PCR Master Mix购自上海翊圣生物科技有限公司;无内毒素质粒小提试剂框购自天根生化科技(北京)有限公司;DNA纯化试剂框AxyPrepTM PCR Cleanup Kit购自美国AXYGEN公司;B2M-PE单克隆抗体(#A15770)、PD-1-PE单克隆抗体(#12-9985-82)购自美国Invitrogen公司;Ficoll-PaqueTM PLUS单核细胞分离液购自美国GE Healthcare公司;Lipofectamine2000、CD3磁珠、CD3/CD28磁珠购自美国Invitrogen公司;Lonza Cell Line Nucleofector Kit V(#amaxa-V4XP-30)电穿孔试剂框、X-VIVO15培养基购自瑞士LONZA公司;无血清培养基Opti-MEM、双抗(1%青霉素-链霉素)购自美国Gibco公司;胎牛血清(fetal bovine serum,FBS)、0.25%胰蛋白酶、磷酸盐缓冲液(phosphate-buffer saline,PBS)、RPMI 1640培养基购自美国Corning公司,DMEM高糖培养基购自美国HyClone公司;嘌呤霉素(puromycin, PM)购自德国Qiagen公司;氨苄青霉素(ampicillin,AMP)、50×TAE电泳缓冲液等试剂购自生工生物(上海)工程股份有限公司。
1.2 初筛sgRNA
1.2.1 sgRNA的设计
利用CCTop-CRISPR/Cas9 target online predictor位点针对B2M和PD-1基因各设计2条sgRNA;针对B2M sgRNA及PD-1 sgRNA各设计一对体外扩增(PCR)引物:B2M sg1-2 F: ATTCCTGAAGCTGACAGCAT;B2M sg1-2 R: TTATCGACGCCCTAAACTTTGT;PD-1 sg1-2 F: AGAAGCTGCAGCCTCACGTAGAAG;PD-1 sg1-2 R: AGAGGTAGGTGCCGCTGTCATTG;pGL3-U6-sgRNA-PGK-puromycin质粒Sanger测序引物Assembly F: CGATTAGTGAACGGATCTCGACG。在NCBI位点验证其特异性。Sanger测序由苏州金唯智生物科技有限公司完成。
1.2.2 质粒构建
1.2.2.1 载体线性化
将带有U6启动子的pGL3-U6-PGK-puromycin质粒载体用高保真限制性内切酶BsaⅠ-HF酶切消化(37℃水浴过夜);酶切产物用AxyPrepTM PCR Clean up Kit试剂框纯化,用超微量分光光度仪(Nanodrop Spectrophotometer)测浓度并标记,-20℃冰箱保存。
1.2.2.2 退火及连接
将依据sgRNA序列合成的寡聚核苷酸(Oligo)成对变性退火形成双链:取正向和反向Oligo各1 μl,按说明书加NEBuffer 2(#B7002S)进行退火,条件:95℃ 5 min;95℃ 30s;85℃ 30 s,每个循环降2℃,30个循环;25℃ 1 min,每个循环降0.1℃,10个循环。退火产物1 μl加线性化载体(pGL3-U6-PGK-puromycin)50 ng、Solution Ⅰ(DNA Ligation Kit Ver 2. 1)1.5 μl,混匀,16℃连接30 min。
1.2.2.3 转化、验证并抽提质粒
连接产物转化至DH5&α;,涂AMP抗性(终浓度100 μg/ml)LB板,培养箱(37℃、5%CO2)过夜培养(12~16 h),挑取数个单菌落于含有AMP(终浓度100 μg/ml)的LB培养基中,培养箱(37℃、5%CO2)培养1~2 h,取2 μl菌液加10 μl 2×Hieff PCR Master Mix、Assembly F 1 μl、反向B2M sgRNA1/2或反向PD-1 sgRNA1/2 1 μl,加水补足体积至20 μl,置于PCR仪扩增,条件:95℃ 5 min;95℃ 30 s,62℃ 30 s,每个循环降0.2℃,72℃ 30 s,30个循环;72℃ 5 min。PCR产物凝胶电泳,选取能扩增出单一明亮且位置正确的条带所对应的菌液送测序;将经测序鉴定正确连接的菌液接种于10~15 ml含AMP的LB培养基,摇菌(37℃,250 r/min,12~16 h),用天根无内毒素质粒小提试剂框抽提质粒pGL3-U6-sgRNA-PGK-puromycin,测浓度并标记后置于-20℃冰箱保存。
1.2.3 Lipofectamine2000转染HEK293T细胞筛选高效率sgRNA
将生长状态良好的HEK293T细胞消化离心后,用含10% FBS的DMEM高糖培养基重悬细胞,以2×105个细胞/毫升密度铺满24孔板(500 μl细胞悬液每孔);按质粒SpCas9:pGL3-U6-sgRNA-PGK-puromycin=2:1比例(共1 μg)加入EP管(A液),取Lipofectamine2000 2 μl于另一EP管(B液),分别加50 μl无血清培养基Opti-MEM稀释混匀并室温静置5 min,将B液加入A液中,混匀后室温静置20 min,缓慢滴入HEK293T细胞中(实验组),对照组HEK293T细胞不加含sgRNA的质粒部分,余条件与实验组保持相同。6 h后更换为新鲜完全培养基(DMEM+10%FBS+1%双抗),24 h加PM(终浓度2 μg/ml),72 h收集细胞提取基因组DNA,分别对应用B2M sg1-2 F/R及PD-1 sg1-2 F/R引物扩增相应sgRNA附近片段(根据Phanta Max Super-Fidelity DNA Polymerase试剂说明书),扩增产物送测序。
1.3 T细胞的分离、分选与活化
T细胞是从新鲜的全血中分离得到。全血来自于健康献血者。使用淋巴细胞分离液(Ficoll-PaqueTM PLUS)从全血中分离得到外周血单个核细胞(peripheral blood mononuclear cells,PBMC),重悬于RPMI 1640培养基中。利用CD3磁珠分选出CD3+T细胞;用CD3/CD28磁珠刺激活化T细胞,以含10%FBS及白细胞介素(IL-2、IL-7、IL-15)的X-VIVO15 T细胞完全培养基按照1×106个细胞/毫升密度置于无菌培养箱(37℃、5%CO2)进行培养。
1.4 sgRNA在T细胞中的效率验证
生长状态良好的T细胞计数后分组(2×106个细胞/份),离心(300 g,8 min)后弃上清液。以Nucleofector Solution:Supplement=82:18比例配制电穿孔缓冲液2×20 μl,用缓冲液重悬T细胞;按照sgRNA(B2M sgRNA1或PD-1 sgRNA1):Cas9蛋白=150 pmol:30 pmol比例轻轻混匀,形成RNP复合物,吸取T细胞悬液,加入RNP复合物,将混合物与T细胞(实验组)混匀后轻轻转至电穿孔杯中,置于电穿孔仪进行电穿孔,往电穿孔杯加入预热的培养基,对照组细胞不加sgRNA,余条件与实验组保持相同。将细胞转入预先铺有预热的培养基的孔板中,无菌培养箱(37℃、5%CO2)培养48 h后分别收取实验组及对照组细胞,提取基因组DNA并行PCR分析,产物送测序;纯化PCR产物(AxyPrep PCR Clean up Kit试剂框)后进行TA克隆:取1 μg纯化PCR产物加5 μl 10×A-tailing Buffer、4 μl dNTP mix、0.5 μl A-tailing Enzyme,加水补足至50 μl体系,72℃金属浴20 min,冰置2 min;取加A尾产物2 μl加pMDTM19-T Vector 0.5 μl、SolutionⅠ 2.5 μl,16℃,≥1 h(PCR仪)。已连接T载体的产物转化至DH5&α;,将转化菌涂AMP抗性板,培养箱培养过夜(12~16 h)。每板挑取25个单菌落,分别接种于LB培养基(含AMP),菌液2 μl加通用引物M13-47 FOR及M13-48 REV各1 μl、2×Hieff PCR Master Mix 10 μl,加水补足体积至20 μl,进行菌液PCR,电泳,选择单一明亮且位置正确的条带所对应菌液送测序。
1.5 构建双基因敲除的T细胞
按照B2M sgRNA1:PD-1 sgRNA1:Cas9蛋白=150 pmol:150 pmol:30 pmol比例配制电穿孔缓冲液100 μl(实验组);对照组不加sgRNA,其他条件与实验组保持相同。余按照1.4中方法电穿孔T细胞后加培养基铺6孔板。培养箱(37℃、5%CO2)培养48 h,分别收取适量细胞提取基因组DNA,用B2M sg1-2 F/R和PD-1 sg1-2 F/R引物进行PCR并送测序;分别进行TA克隆;电穿孔后96 h收取两份实验组细胞、三份对照组细胞(5×106个细胞/份样品),双敲除组及对照组细胞各2份分别避光孵育单克隆抗体(B2M-PE & PD-1-PE)15~20 min,PBS洗去背景抗体后重悬细胞,上流式细胞仪检测。B2M或PD-1基因表达下调率=(对照组表达量-实验组表达量)/对照组表达量×100%。
2
结果
2.1 sgRNA的设计与构建
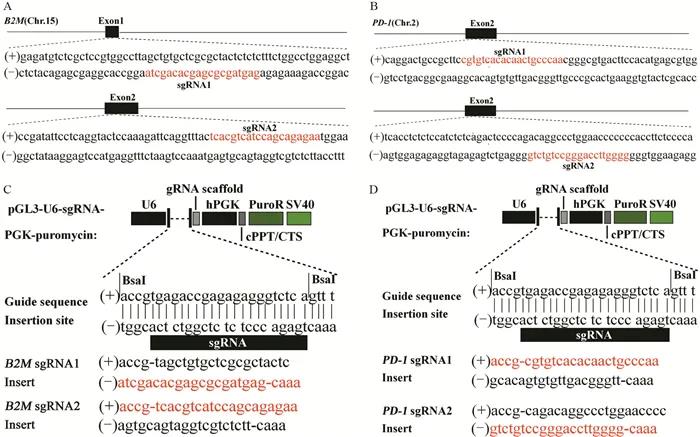
为获得B2M及PD-1基因的移码突变,针对B2M基因1号及2号外显子分别设计一对sgRNA,见图 1A;针对PD-1基因1号外显子设计两对sgRNA,见图 1B。将订购的单链sgRNA上下游退火形成双链,载体质粒pGL3-U6-PGK-puromycin含有U6启动子、BsaⅠ-HF特异性酶切位点、PM及AMP抗性标签,经高保真限制性内切酶BsaⅠ-HF酶切后形成黏性互补末端,与双链sgRNA连接形成pGL3-U6-PGK-sgRNA-puromycin质粒后转化至DH5&α;,涂板,隔天挑取单克隆测序,选择含构建成功质粒的菌液扩大培养并抽提质粒,见图 1C、1D。
|
sgRNAs are both in red; A: schematic diagrams of B2M sgRNAs targeting sites, sgRNA1 and sgRNA2 were located in exon 1 and exon 2 of B2M gene, respectively; B: schematic diagrams of PD-1 sgRNAs targeting sites, sgRNA1 and sgRNA2 were both located in exon 2 of PD-1 gene; C: plasmid skeletal structure of pGL3-U6-sgRNA-PGK-puromycin and the insert sequences of B2M sgRNAs; D: plasmid skeletal structure of pGL3-U6-sgRNA-PGK-puromycin and the insert sequences of PD-1 sgRNAs. |
图 1 sgRNA的设计与构建 |
2.2 sgRNA在HEK293T细胞中的筛选
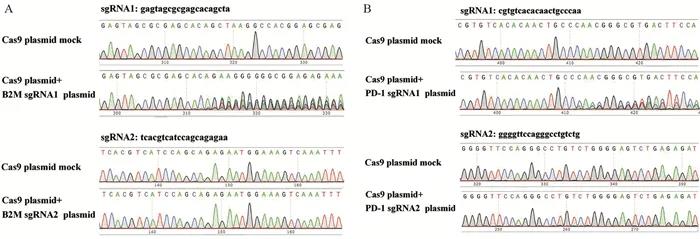
利用Lipofectamine 2000脂质体转染方法将构建好的质粒pGL3-U6-sgRNA-PGK-puromycin与spCas9质粒共转染HEK293T细胞。转染后24 h加入PM(终浓度2 μg/ml),72 h后对照组细胞全部死亡,实验组约5%细胞死亡。用PBS轻洗实验组细胞,去掉死亡细胞后余下细胞计数,实验组及对照组各取5~10万个细胞提取基因组DNA,并体外扩增B2M及PD-1基因组sgRNA附近序列,Sanger测序结果显示B2M sgRNA1及PD-1 sgRNA1具有高编辑效率(约90%),见图 2。
|
Cas9 plasmid mock: control group; Cas9 plasmid + sgRNA plasmid: experimental group. A: Sanger sequencing of B2M sgRNA1 and B2M sgRNA2 parts in HEK293T cells; B: Sanger sequencing of PD-1 sgRNA1 and PD-1 sgRNA2 parts in HEK293T cells. |
图 2 B2M sgRNA和PD-1 sgRNA在HEK293T细胞中的筛选 |
2.3 初筛的高效率sgRNA在T细胞中的效率验证
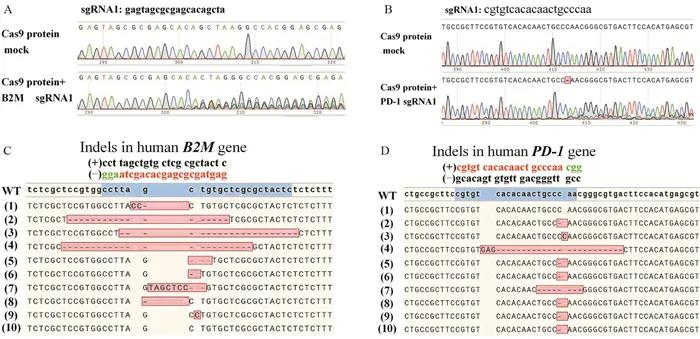
将B2M sgRNA1、PD-1 sgRNA1分别与Cas9蛋白混合形成RNP复合物,使用LONZA电穿孔试剂框分别电穿孔T细胞;48 h后收集细胞提取基因组DNA,分别用B2M sg1-2 F/R和PD-1 sg1-2 F/R引物进行PCR并对扩增产物进行测序,Sanger测序结果表明B2M sgRNA1及PD-1 sgRNA1在T细胞中同样具有高编辑效率,见图 3A、3B。将B2M和PD-1基因编辑后的T细胞分别进行裂解,提取基因组DNA进行PCR,PCR产物纯化并克隆至T载体,TA克隆测序结果分析显示B2M sgRNA1和PD-1 sgRNA1在T细胞中的编辑效率均达到90%,见图 3C、3D。
|
Cas9 protein mock: control group; Cas9 protein+sgRNA: experimental group. A: the result of Sanger sequencing of B2M sgRNA1 in B2M knock-out T cells; B: the result of Sanger sequencing of PD-1 sgRNA1 in PD-1 knock-out T cells; C: TA cloning of B2M sgRNA1 in B2M knock-out T cells; D: TA cloning of PD-1 sgRNA1 in PD-1 knock-out T cells. |
图 3 B2M sgRNA1或PD-1 sgRNA1编辑效率在T细胞中的验证 |
2.4 构建B2M及PD-1双基因敲除的T细胞
经验证的高效sgRNA(B2M sgRNA1和PD-1 sgRNA1)与Cas9蛋白混合形成RNP复合物并电穿孔T细胞。Cas9 RNP方式电穿孔T细胞后的基因修饰通常在转染后48 h内完成,但丢失的靶蛋白表达所需时间是可变的,既往对于T细胞中多个靶标进行流式分析或Western Blot显示通常48~72 h足以看到靶蛋白表达的完全丧失。故而我们在电穿孔后48 h后收取5~10万个细胞提取基因组DNA进行PCR,测序并进行TA克隆;72 h后收集实验组、对照组T细胞分别孵育B2M-PE及PD-1-PE单抗并上机进行流式细胞术分析蛋白表达情况。
Cas9结合sgRNA形成复合体,通过与靶序列以碱基互补配对原则定位至特定部位,引发特异性的切割靶序列导致双链DNA断裂(double-strand DNA breaks,DSBs),从而引起生物体内DNA的损伤修复机制,包括同源定向重组(homology-directed recombination,HDR)和容易出现错配的非同源末端连接(non-homologous end joining,NHEJ)。出现错配的NHEJ事件后最终产生碱基替换、插入或缺失等不同类型基因修饰,从而达到编辑基因的效果。实验组中Sanger测序峰值图中的双峰或多峰显示了在B2M和PD-1基因座处CRISPR介导的NHEJ事件,见图 4A。TA克隆测序分析显示多种类型的编辑结果主要集中在sgRNA位置附近,包括单/多碱基替换、敲除及插入,B2M及PD-1基因层面编辑效率均高达90%,见图 4B。
|
A: Sanger sequencing showed CRISPR-mediated NHEJ events in B2M and PD-1 genes in double genes knock-out T cells; B: Insertion and deletion were observed by PCR-amplified TA cloning sequence analysis following Cas9-mediated B2M and PD-1 genes cleavage; C: FACS detected B2M and PD-1 protein expression levels in double genes knock-out T cells. Negtive: Control group(unhatched antibody); Cas9 protein mock: control group(incubated antibody); Cas9 protein+B2M sgRNA1+PD-1 sgRNA1: experimental group. |
图 4 B2M和PD-1双基因敲除的T细胞中的编辑效率 |
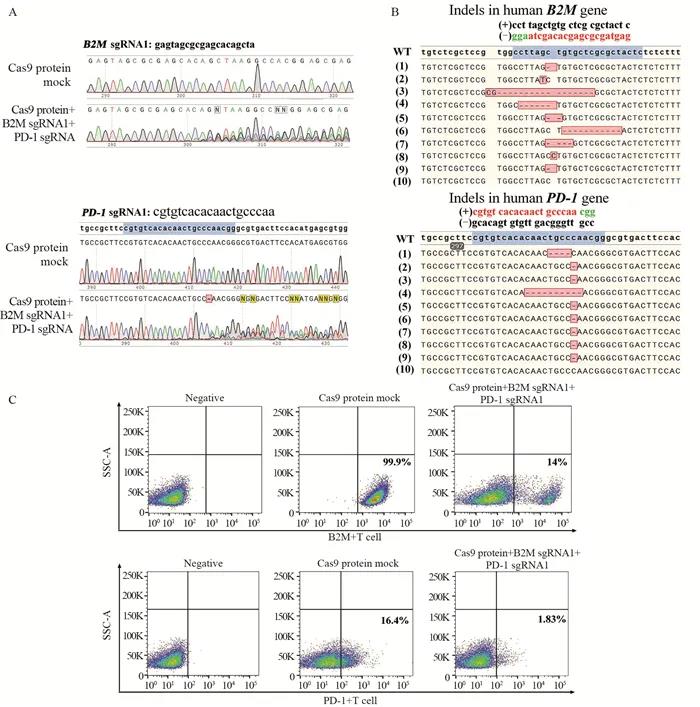
在T细胞中,B2M蛋白为组成性表达,而PD-1主要由TCR介导T细胞活化从而被诱导表达,高表达于衰竭T细胞[8]。流式分析对照组T细胞中B2M及PD-1蛋白表达量分别为99.9%和16.4%,而实验组分别为14%、1.83%,表达下调率分别达86%、89%,蛋白层面同样显示编辑效果显著,见图 4C。
3
讨论
来源于微生物(细菌和古菌)的CRISPR/Cas9因操作简单、高效、安全性高和脱靶率低等特点,一经发现便被广泛采用,已经成为最常用的可编辑核酸酶。Cas9系统由DNA内切酶Cas9蛋白和sgRNA两部分组成,可以诱导人和小鼠等多个物种细胞内源基因组位点的精确切割[9-10],从而使原代T细胞的体外编辑成为可能。但由于缺乏有效的转染方法,由Cas9系统所介导的人原代T细胞内源基因修饰受到限制。传统病毒传递[11-12]或电穿孔转入构建体质粒方式[13-14]在T细胞中靶向编辑效率相对低下,且后者细胞毒性大,常规T细胞存活率低于50%。为了解决这一困境,多个学者尝试利用Cas9 RNP方式电穿孔人原代T细胞,通过不断探索优化实验方案,发现靶基因的编辑效率可达50%~90%[15-17]。研究证实通过Cas9 RNP方式编辑后的T细胞的活性及功能并未受到影响,总体T细胞存活率约60%~80%,并且单次转入RNP总量的增加并不降低T细胞活性及IFN-&γ;等细胞因子的分泌功能[18],较传统电穿孔导入构建体质粒方式显著降低T细胞的毒性。此外,由于Cas9 RNP复合物在细胞内快速降解,其脱靶效应远低于既往的常规基因递送方法[19]。
近年来,陆续有研究探讨利用不同组合模式的Cas9系统(SpCas9质粒、Cas9 mRNA或Cas9蛋白等与不同sgRNAs)对T细胞进行PD-1或B2M基因敲除,以改善或优化其功效。Seki等[18]通过Cas9 RNP技术敲除鼠和人原代T细胞中PD-1等多个靶基因的效率超过85%,且PD-1基因敲除并不影响T细胞的活化及功能。Rupp等[5]通过体内外实验研究证实PD-1基因敲除显著增强CAR T细胞在体外的肿瘤细胞杀伤效应,同时增强了其对体内PD-L1+肿瘤异种移植物的清除能力。胡边等[20]发现,PD-1基因敲除并不影响CAR的表达。另一项研究[21]通过Cas9 mRNA与sgRNA组合电转T细胞以实现B2M基因敲除,结果显示B2M及HLA-Ⅰ类分子双阴性比例达79.9%,而T细胞中B2M基因的缺失显著降低了其对共培养同种异体PBMC的反应性;该研究[21]同时还证实T细胞内B2M及PD-1基因的敲除并不影响CAR T细胞的体内植入、增殖及抗肿瘤功效。然而,上述研究虽然已经显著提高了T细胞内多个基因的编辑效率,但是仍无法实现目的基因在种族水平上的完全敲除。为了消除“通用”T细胞产品的同种异体移植免疫排斥反应并增强T细胞功能,我们设计并进行了此次实验研究:首先利用HEK293T细胞完成sgRNA编辑效率的初步筛选,然后分别进行T细胞中B2M及PD-1单基因敲除以再次验证sgRNA效率,最后通过同时加入2种高效sgRNA与Cas9蛋白结合形成RNP复合物,用电穿孔方式转入T细胞中进行双基因敲除。在未进行任何纯化或筛选的情况下,双基因敲除T细胞在基因层面B2M及PD-1的编辑效率均达到了90%,蛋白表达下调率分别为86%、89%。
总之,我们首次采用Cas9 RNP技术实现B2M及PD-1双基因高效率同步敲除。提供了一种更高效率的T细胞基因编辑实验流程,克服了通过病毒或一体化载体质粒构建方式编辑T细胞内源基因的局限性。在显著提高编辑效率的同时,该技术极大程度简化了实验步骤,节约了时间及经济成本。今后,我们将通过体外和体内实验检测已被基因编辑的T细胞的增殖、免疫功能等生物学特征及脱靶效率,并验证是否能达到消除“通用”T细胞的同种异体移植排斥反应并增强其功能的目的。此外,我们拟通过优化实验条件,优化sgRNA设计方案,增加针对多个靶基因的多个sgRNA并调整Cas9蛋白与sgRNA比例进行不同组合,以实现同步编辑T细胞多内源基因(例如T细胞其他抑制性位点,包括细胞毒性T细胞抗原-4(cytotoxic T cell antigen-4,CTLA-4)、T细胞免疫球蛋白黏蛋白域-3(T cell immunoglobulin and mucin domain-3,TIM-3)、淋巴细胞激活基因-3(lymphocyte activation gene 3,LAG-3)等),并进一步提高编辑效率,降低T细胞毒性。我们相信,将Cas9系统介导的基因编辑技术与非HLA依赖的CAR T细胞的慢病毒转导技术相结合,有望在克服同种异体“通用”CAR T细胞的免疫排斥问题同时增强T细胞功能,拓宽CAR T细胞的发展前景,为肿瘤医生及患者提供更多高效的治疗选择方案。
徐娅1, 付烊2,朱诗聪3,章必成3
作者单位:1. 430065 武汉,武汉科技大学医学院;2. 441001 襄阳,湖北中医药大学附属襄阳医院肿瘤科;3. 430060 武汉,武汉大学人民医院肿瘤中心
基金项目: 湖北省自然科学基金(2019CFB635);湖北省卫健委科研课题(WJ2019M194);希思科-恒瑞肿瘤研究基金(Y-HR2018-328);希思科-BMS肿瘤免疫治疗研究基金(Y-BMS2019-003)
2020,47(06):403-410. DOI:10.3971/j.issn.1000-8578.2020.19.1603
[1] Ren J, Zhang X, Liu X, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation[J]. Oncotarget, 2017, 8(10): 17002-17011. DOI:10.18632/oncotarget.15218
[2] Torikai H, Cooper LJ. 转录 implications for off-the-shelf immune cells expressing chimeric antigen receptors[J]. Mol Ther, 2016, 24(7): 1178-1186. DOI:10.1038/mt.2016.106
[3] Shao L, Zhang Y, Pan X, et al. Knockout of β-2 microglobulin enhances cardiac repair by modulating exosome imprinting and inhibiting stem cell-induced immune rejection[J]. Cell Mol Life Sci, 2020, 77(5): 937-952. DOI:10.1007/s00018-019-03220-3
[4] Wang D, Quan Y, Yan Q, et al. Targeted disruption of the β2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells[J]. Stem Cells Transl Med, 2015, 4(10): 1234-1245. DOI:10.5966/sctm.2015-0049
[5] Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells[J]. Sci Rep, 2017, 7(1): 737. DOI:10.1038/s41598-017-00462-8
[6] Ouchi Y, Patil A, Tamura Y, et al. Generation of tumor antigen-specific murine CD8+ T cells with enhanced anti-tumor activity via highly efficient CRISPR/Cas9 genome editing[J]. Int Immunol, 2018, 30(4): 141-154. DOI:10.1093/intimm/dxy006
[7] John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells[J]. Clin Cancer Res, 2013, 19(20): 5636-5646. DOI:10.1158/1078-0432.CCR-13-0458
[8] Okada M, Chikuma S, Kondo T, et al. Blockage of core fucosylation reduces cell-surface expression of PD-1 and promotes anti-tumor immune responses of T cells[J]. Cell Rep, 2017, 20(5): 1017-1028. DOI:10.1016/j.celrep.2017.07.027
[9] Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems[J]. Science, 2013, 339(6121): 819-823. DOI:10.1126/science.1231143
[10] Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering[J]. Cell, 2014, 157(6): 1262-1278. DOI:10.1016/j.cell.2014.05.010
[11] Li C, Guan X, Du T, et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9[J]. J Gen Virol, 2015, 96(8): 2381-2393. DOI:10.1099/vir.0.000139
[12] Wang W, Ye C, Liu J, et al. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection[J]. PLoS One, 2014, 9(12): e115987. DOI:10.1371/journal.pone.0115987
[13] Su S, Hu B, Shao J, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients[J]. Sci Rep, 2016, 6(1): 20070.
[14] Mandal PK, Ferreira LM, Collins R, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9[J]. Cell Stem Cell, 2014, 15(5): 643-652. DOI:10.1016/j.stem.2014.10.004
[15] Schumann K, Lin S, Boyer E, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins[J]. Proc Natl Acad Sci U S A, 2015, 112(33): 10437-10442. DOI:10.1073/pnas.1512503112
[16] Hendel A, Bak RO, Clark JT, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells[J]. Nat Biotechnol, 2015, 33(9): 985-989. DOI:10.1038/nbt.3290
[17] Gomes-Silva D, Srinivasan M, Sharma S, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies[J]. Blood, 2017, 130(3): 285-296. DOI:10.1182/blood-2017-01-761320
[18] Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells[J]. J Exp Med, 2018, 215(3): 985-997. DOI:10.1084/jem.20171626
[19] Liang X, Potter J, Kumar S, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection[J]. J Biotechnol, 2015, 208: 44-53. DOI:10.1016/j.jbiotec.2015.04.024
[20] 胡边. CRISPR-Cas9介导的人原代T细胞的基因编辑[D].南京: 南京大学, 2018. [Hu B. CRISPR-Cas9 mediated gene editing in primary human T cells[D]. Nanjing: Nanjing University, 2018.]
[21] Ren J, Liu X, Fang C, et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition[J]. Clin Cancer Res, 2017, 23(9): 2255-2266. DOI:10.1158/1078-0432.CCR-16-1300
