为何肺癌靶向治疗这么难?你可能需要了解这个信号通路!
EGFR-TKI 治疗非小细胞肺癌(NSCLC),耐药不可避免。MET 基因变异作为一种 EGFR TKIs 耐药机制的同时,也可以孤立驱动肺癌的发生和发展。而目前 NCCN 等指南已将 MET 作为检测靶点。
2020 年最新版(2020.V6)非小细胞肺癌 NCCN 指南推荐,肺癌患者应检测传统靶点有:EGFR、ALK、ROS1、BRAF、KRAS、NTRK、PD-L1;建议检测的新兴靶点有:MET、RET。

图1:2020 年最新版(2020.V6)非小细胞肺癌
NCCN 指南对检测靶点的推荐
肺癌中驱动癌变的关键信号
——MET 基因变异的分子机制
MET 信号通路的异常激活促进了肺癌细胞的存活、增殖、侵袭以及对 EGFR 靶向药物的耐药。MET 通路异常激活的形式主要有 MET 过表达、重排、扩增、MET 14 号外显子突变。
MET 过表达是指 MET 蛋白的表达增多,而不伴有 MET 基因的扩增或突变。这种过表达引起了不依赖配体的 MET 通路磷酸化和下游信号传导通路的活化。
MET 重排是在二十世纪 90 年代首次发现的。研究人员发现色氨酸(TRP)基因与 MET 基因发生重排,此后在 NSCLC 中发现了 MET 与包括 KIF5B、 CD47 等其他基因的重排。
MET 基因扩增是指 MET 基因拷贝数(GCN)增加。这可能与通过断裂-融合-桥连机制(breakage-fusion-bridge mechanisms)引起的基因局部复制有关。较高的 GCN 也可以继发于 7 号染色体的多体症。MET 扩增通过 MET 蛋白质过表达和激酶的持续激活引起 MET 信号通路失调。
MET 14 号外显子突变是孤立的肿瘤驱动因子。MET 14 号外显子编码的近膜结构域是关键的负性调控区,包含 E3 泛素连接酶 c-CbI 酪氨酸结合位点(Y1003)(图1)。正常情况下,MET 14 号外显子侧翼的内含子在前 mRNA 中被剪接出来,使得含MET 14 号外显子的 mRNA 被正常翻译。在 MET 14 号外显子上游端和下游端的剪接位点可能发生包括碱基对的点突变、删除、插入或复杂突变(插入缺失)等突变,它们都会影响 14 号外显子周围内含子剪接连接点的保守序列,这些突变可能导致前 mRNA 的剪接过程中 14 号外显子随其两端的内含子一同被剪接,进而导致 MET 14 号外显子缺失。MET 14 号外显子编码的关键负性调控区域缺失将导致 c-Met 蛋白泛素化和降解受阻,引起下游信号的持续激活,最终促进肿瘤的发生[1]。
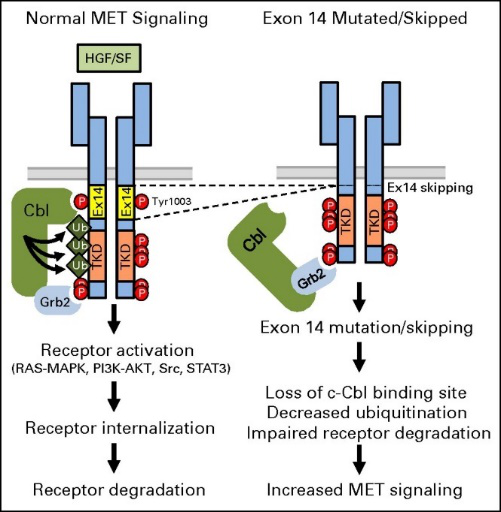
图2:MET 14 号外显子
跳跃突变的发生机制示意图[1]
各种 MET 基因变异的发生率
MET 过表达、重排、扩增、MET 14 号外显子突变这几种 MET 通路改变的发生频率各有不同,检测方法也不尽相同。
MET 过表达的发生频率范围介于 22%~75% 之间[2]。MET 过表达被认为是预后不良的因素,与死亡风险增加显著相关(HR = 1.52),且 IHC 检测出的 MET 阳性与总生存(OS)降低显著相关(HR = 1.55)[2]。
MET 重排的发生率未知,有研究报道 2410 例 NSCLC 患者中 MET-ATXN7L1 重排发生率约为 0.04%[2]。
在 NSCLC 中,原发 MET 扩增的发生频率为 1%~5%[2]。其发生频率与样本选择、检测方法和 MET 扩增阳性的阈值设定有关。原发 MET 扩增在低分化预后不良的腺癌中发生更频繁,且 MET 扩增与较短的 OS 显著相关。继发 MET 扩增是 EGFR TKI 耐药的主要机制之一,约占 EGFR TKI 耐药机制的 5%~20%[2]。目前,Southern 印迹、PCR,以及荧光原位杂交技术(FISH)可用于分析 MET 基因扩增 [1]。
MET 14 号外显子跳跃突变是近年来新的研究热点。根据 NGS 分析,在所有 NSCLC 中 MET 14 号外显子跳跃突变发生率为 1.7%~4.3%[2],而在恶性程度高、预后极差的肺肉瘤样癌中,其突变率高达 31.8%[2]。研究显示 MET 14 号外显子跳跃突变与 EGFR/ALK 等一样,是一种能够孤立致癌的驱动基因,且 MET 14 号外显子跳跃突变也是一项孤立的预后不良指标,若同时也伴有高水平 MET 扩增则可预测较差的生存率。由于 MET 14 号外显子缺失的突变具有高度多样性,这也使其分子诊断存在挑战。基于 NGS 技术的 RNA 测序能够对 MET 14 号外显子跳跃突变进行更为准确的检测,可以作为 DNA 测序的补充。
抗 MET 疗法如何实现?各路「英雄」争伯仲
MET 通路可以通过几种不同机制进行靶向。根据 HGF/c-Met 信号通路中作用位点的不同,可将 MET 靶向药物分为三大类:抗 HGF 单克隆抗体、抗 c-Met 单克隆抗体和小分子 TKI。前两者分别在细胞外与 HGF 和 c-Met 结合,从而阻止 HGF 与 c-Met 的结合及受体磷酸化,阻止信号传导;小分子 TKI 与 MET 基因编码的酪氨酸激酶结构域中与细胞内 ATP 结合的位点结合,阻止磷酸基团传递从而阻止蛋白磷酸化,阻断 MET 通路信号传导,从而间接抑制由 MET 通路激活导致的肿瘤细胞增殖、转移及侵袭。此外还有 MET 下游通路拮抗剂,可拮抗 MET 通路下游分子如 mTOR、MEK 等分子从而抑制信号转导。
目前研究最多的也最具有治疗潜力的是小分子 TKI。根据构象和作用机制,TKI 可以分为三种类型:I 型 TKI 是 ATP 竞争性的,与 MET 主链中的氨基酸残基形成氢键,其中又分为 Ia 型和 Ib 型,区别在于 Ib 型 TKI 可以结合的位点较少,不包括甘氨酸残基的 G1163 位点,因此特异性较高;II 型 MET-TKI 一般为多靶点 TKI,不仅占据 ATP 结合位点,还能通过管家基因(即所有细胞中均要稳定表达的基因)突变进入非活性 DFG-out 构象形成的疏水口袋,对产生二次突变的 MET 仍具有抑制作用,故或许可以逆转由 Y1230 等位点突变引起的 I 型 MET-TKI 耐药;III 型 MET-TKI 作用于与 ATP 结合位点完全不同的变构位点,目前尚没有这类变构抑制剂药物进入临床研究阶段[3]。
总结
MET 基因变异以 MET 过表达、重排、扩增、MET 14 号外显子突变这几种形式为主,具有重要的临床意义。针对四种异常通路,多种靶向药物争相上场,主要包括针对 c-MET/HGF 单克隆抗体及小分子 MET-TKI。MET-TKI 是其中最具潜力的药物,已显示出颇具希望的初步疗效。对于 MET 14 号外显子跳跃突变患者,或许曙光即将到来。
参考文献
[1] Awad MM. Impaired c-Met Receptor Degradation Mediated by MET Exon 14 Mutations in Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34(8): 879-881.
[2] Bylicki O, et al. Targeting the MET-Signaling Pathway in Non-Small-Cell Lung Cancer: Evidence to Date. Onco Targets Ther. 2020;135691-5706.
[3] 韩森, 马旭, 方健. 非小细胞肺癌MET基因突变的机制及靶向药物研究进展. 中国肺癌杂志. 2020;23(7):609-614.
CN-64462
学术专业文章仅供医疗专业人士参考
内容审核:刘晔 梁思
题图来源:站酷海洛
