鱼鳞病
寻常型鱼鳞病(Ichthyosis Vulgaris)又称干皮病(xeroderma)、单纯型鱼鳞病(ichthyosis simplex)、光泽鱼鳞病(ichthyosis nitida),为具有不全外显的常染色体显性遗传病,是一种临床常见病,多发病。 寻常型鱼鳞病的特征是缺乏透明角质颗粒的组成成分丝聚合蛋白原。目前认为这是与丝聚合蛋白基因(FLG基因)缺失有关。
寻常型鱼鳞病的发病机制
发病机制:与皮肤中细丝聚合蛋白基因(filaggrin,FLG基因)缺失有关。FLG是皮肤表皮角质层角蛋白包膜的重要成分。这些活性聚丝蛋白肽协助角蛋白丝聚集成密集的角蛋白束,形成紧密的蜂窝状态和鳞状结构。然后在谷氨酰胺转胺酶的作用下形成角蛋白包膜和皮肤屏障。聚丝蛋白肽帮助角蛋白丝聚合,因此得名。
角蛋白包膜形成过程中,聚丝蛋白最终从角蛋白束中分离出来,进一步降解为亲水游离氨基酸和吸水衍生物,帮助皮肤储存水分,维持皮肤水分。它的代谢产物作为一种渗透物质,促进水渗透到角质层,所以角质层将有很大一部分的含水量。
聚丝蛋白的一系列代谢过程表明,FLG的缺失与皮肤干燥有关。
同时也有研究表明,FLG基因也是皮炎、哮喘、过敏性鼻炎的易感基因。
寻常性鱼鳞病的症状
出生时无症状,常在婴幼儿发病。
于背及四肢伸侧出现淡褐至深褐色菱形或多角形鳞屑,紧贴在皮肤上面,其边缘呈游离状,对称分布。下肢尤甚。
严重时可波及躯干、四肢屈侧等部位,腋窝、臀裂常不累及。
头皮可有轻度糠状鳞屑。
手背常见毛囊性角化丘疹,掌跖常见线状皲裂和掌纹加深。损害轻重不等,一般无自觉症状。
病情与季节关系密切。夏季轻,冬季重。部分患者可有遗传过敏性疾病的个人史及家族史,如湿疹、鼻炎和哮喘。病程不一致,一些患者到成年后。病情改善。
寻常型鱼鳞病的治疗
口服大剂量维生素A或维生素A酸有一定疗效。外用10%尿素软膏、0.1%维生素A酸软膏等可减轻皮损。病情可在温暖、潮湿的气候中缓解。
外用油脂霜剂可有一些效果,10%尿素软膏的效果较佳。0.1%维A酸霜或油膏对此型皮损无效。盐水浴可通过盐水与角质层作用而利于本病。40%~60%丙二醇水溶液、3%~10%乳酸及其他α-羟基软膏亦有一定疗效。
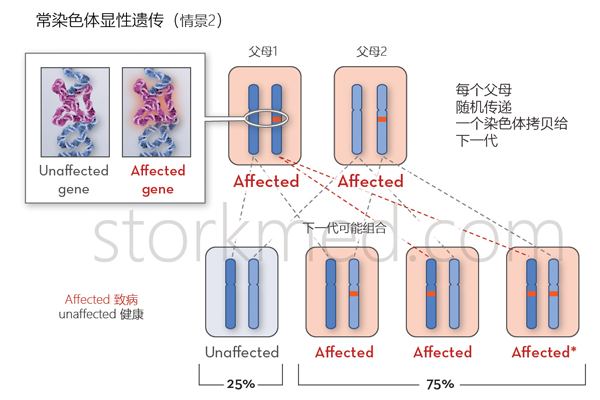
寻常型鱼鳞病的遗传模式
寻常型鱼鳞病为常染色体显性遗传病,缺陷基因呈显性表达,后代子女都有50%的发病可能性。
寻常型鱼鳞病的生育干预
可以通过FLG基因检测进行遗传病筛查,也为后代的优生优育提供了遗传咨询依据。如果想避免避免将致病基因遗传给下一代,可以通过三代试管婴儿技术,在制备胚胎时有效筛别。
三代试管婴儿技术,在传统试管婴儿基础上,加入了染色体筛查。在胚胎移植前,通过染色检测,筛选出优质胚胎,确保婴儿不再患有染色体疾病(包括鱼鳞病和其他染色体综疾病),从而提升试管成功率。尤其适用于伴侣双方中至少一人患有鱼鳞病(或为该类遗传病的携带者)以及已经生育过鱼鳞病孩子的家庭。
Sjogren-Larsson 综合征(SLS) 是由 ALDH3A2 基因( 609523 )中的纯合或复合杂合突变引起的,该基因编码脂肪醛脱氢酶(FALDH),位于染色体 17p11。
| 点位 | 表型 | 表型 MIM 编号 |
遗产 | 表型 映射键 |
基因/位点 | 基因/基因座 MIM 编号 |
|---|---|---|---|---|---|---|
| 17p11.2 | Sjogren-Larsson综合征 | 270200 | AR | 3 | ALDH3A2 | 609523 |
▼ 说明
------
Sjogren-Larsson 综合征是一种常染色体隐性遗传、儿童早期发病的疾病,其特征是鱼鳞病、智力低下、痉挛性下肢轻瘫、黄斑营养不良和白质脑病。它是由脂肪醛脱氢酶缺乏引起的(Lossos 等人总结,2006 年)。
▼ 临床特点
------
Sjogren-Larsson 综合征的皮肤变化与先天性鱼鳞病样红皮病( 242100 )的皮肤变化相似,尽管已经描述了严重程度的相当大的变化( Goldsmith 等,1971 )。Link 和 Roldan(1958)报告了病例。布鲁梅尔等人(1958)将神经系统疾病称为痉挛性四肢瘫痪。Sjogren(1956)和Sjogren 和 Larsson(1957)提出,他们的 28 个病例都来自于大约 600 年前发生的相同突变,并且瑞典北部大约 1.3% 的人口是该基因的杂合子。大约一半的病例有视网膜色素变性。眼底病变由以下人员讨论吉尔伯特等人(1968)。视网膜上闪烁的白点是特征性的。瘀斑在出生时或出生后不久就存在。大多数患者从不走路。身材往往偏矮。大约一半的患者有癫痫发作。限制脂肪摄入和补充中链甘油三酯可改善临床症状。
雷纳等人(1978)描述了 2 个兄弟和一个姐妹的综合征,该综合征结合了 Sjogren-Larsson 综合征的许多特征,但可能不同。他们回顾了具有 Sjogren-Larsson 综合征表型特征的一组疾病。这种Sjogren-Larsson 综合征有时被称为 T. Sjogren 综合征,以区别于Sjogren-Larsson 综合征(见200400 , 270150),Sjogren-Larsson 综合征由出生于 1899 年的瑞典眼科医生 Henrick Sjogren 描述。
Jagell 和 Linden(1982)研究了 1980 年在瑞典活着的所有 36 名 SLS 患者。轻微或中度角化过度,面部不太明显,在出生时就已经存在,但从未见过火棉胶膜。鱼鳞病在婴儿期发展到它的全部范围。皮肤变化集中在颈部和下腹部以及弯曲处,鳞片通常是黑色的。头发和指甲以及出汗能力不受影响。在所有 30 名接受检查的瑞典 Sjogren-Larsson 综合征患者中,眼底的闪光点是强制性的早期征兆(Jagell 等人,1980 年)。
在挪威北部,Gedde-Dahl 等人(1984)遇到一个家庭,其中 3 名同胞患有与 Sjogren-Larsson 综合征非常相似的鱼鳞病,但没有任何相关的神经系统特征;见270220。
威廉森等人(2000)研究了 15 名已证实脂肪醛脱氢酶缺乏症的Sjogren-Larsson 综合征患者,发现所有患者都患有青少年视网膜黄斑营养不良。患者从 1 至 2 岁开始表现出高度特征性的双侧、闪亮的黄白色视网膜点。点的数量随着年龄的增长而增加。黄斑部异常的程度与鱼鳞病的严重程度或神经系统异常的严重程度无关。高比例的患者表现出额外的眼部体征和症状,特别是明显的畏光。
来自 SLS 患者的培养皮肤成纤维细胞显示出由于缺乏脂肪醇:NAD+ 氧化还原酶而导致的十六醇氧化受损。患者和杂合子的缺陷也可以通过研究白细胞来检测(Rizzo et al., 1987)。里佐等人(1988)研究了培养的皮肤成纤维细胞中的脂质代谢。在标记的棕榈酸酯存在下孵育的完整 SLS 成纤维细胞比正常细胞积累了更多的放射性十六醇,而放射性与其他细胞脂质的结合没有改变。SLS 成纤维细胞的十六醇含量异常升高。里佐等人(1988)表明脂肪醇:NAD + 氧化还原酶,催化十六醇氧化成脂肪酸的酶,在 SLS 成纤维细胞中是缺乏的。平均活性是正常成纤维细胞的 13%。来自 2 个专性杂合子的成纤维细胞具有中等水平的酶活性。在后来的报告中,Rizzo 等人(1989)描述了 8 名患者和 9 名专性杂合子的脂肪醇代谢研究。
洛索斯等人(2006)报道了Rogers 等人先前报道的一个近亲阿拉伯家庭的 6 名 SLS 同胞的随访(1995)。这些同胞的年龄从 16 岁到 36 岁不等。他们都表现出这种疾病的典型特征,但严重程度与年龄没有明显的相关性。尽管有一些证据表明黄斑变性进展,但皮肤和神经系统特征并不进展。脑磁共振波谱(MRS) 显示年长同胞的 1.3 ppm 脂质峰值降低,表明疾病活动度降低。洛索斯等人(2006)建议存在补偿因素来解释具有相同突变的同胞之间的临床变异性。
杰克等人(2015)表征了 9 名患者的视网膜发现,年龄从 3 岁到 23 岁不等,具有 SLS 和 ALDH3A2 突变。所有 9 人都表现出全身性鱼鳞病、痉挛性双瘫、畏光、上眼睑皮肤鱼鳞病和闪亮的黄斑晶体。7 名患者 14 只眼的光学相干断层扫描显示黄斑晶体存在于所有层中,但主要存在于内核层和外丛状层。全视网膜厚度减少22%,内核层减少30%,外核层减少40%。眼底自发荧光(FAF) 和荧光素血管造影(FA) 显示视网膜色素上皮萎缩。用 FAF 成像的所有 4 名患者均显示具有晶体的异质黄斑自发荧光。
▼ 生化特征
------
脂肪醇:NAD+ 氧化还原酶是一种复杂的酶,由 2 种不同的蛋白质组成,这些蛋白质依次催化脂肪醇氧化为脂肪醛,然后氧化为脂肪酸。在旨在确定 SLS 的生化缺陷是前一步脂肪醇脱氢酶(FADH) 还是后一步的研究中,脂肪醛脱氢酶(FALDH),Rizzo 和 Craft(1991)表明 FALDH 是选择性缺陷的,而 FADH普通的。SLS 细胞中 FALDH 缺乏的程度取决于用作底物的脂肪醛。当使用十八醛作为底物时,专性 SLS 杂合子中的 FALDH 活性约为平均正常活性的 50%。
▼ 群体遗传学
------
在瑞典,Jagell 等人(1981)追踪了 41 个家庭的 58 名患者,其中 35 人还活着。在这 58 人中,有 45 人出生在瑞典东北部的禁区。Vasterbotten 县的疾病患病率、杂合子频率和基因频率估计分别为每 100,000 人 8.3、2.0% 和 0.01。
▼ 测绘
------
基于连锁分析和等位基因关联,Pigg 等人(1994)将 SLS 基因定位到 17 号染色体。减数分裂重组表明该基因的侧翼是着丝粒上的 D17S805 和端粒侧的 D17S783、D17S959、D17S842 和 D17S925。与 D17S805 的强等位基因关联表明该突变位于该标记附近。单倍型分析与创始人效应一致,此前家谱证据表明了这种效应。在 7 个不同种族的谱系中,Rogers 等人(1995)证实了 SLS 与 17 号染色体的中心区域的连锁。来自 2 个近亲埃及家庭的患者在该区域的所有 9 个标记基因座上都是纯合的,这表明在这些患者中,携带 SLS 基因的 17 号染色体区域在血统上是相同的。作者还确定了几个包含 FALDH 基因和与 SLS 密切相关的 D17S805 标记的 YAC。他们得出结论,FALDH 可能是近端 17p 上醛脱氢酶基因簇的一部分,因为发现对应到 17p11.2的醛脱氢酶基因(ALDH3; 100660 ) 与 FALDH 基因和 D17S805 在 2 个 YAC 上共定位。罗杰斯等人(1995) 在阿拉伯、欧洲混血、美洲原住民和瑞典血统的家庭中发现 SLS 基因座与 17p 的联系,从而为遗传同质性提供了证据。
▼ 分子遗传学
------
通过对来自 3 名无关 SLS 患者的 FALDH 基因的序列分析,Rogers 等人(1997)鉴定了不同的突变(参见,例如,609523.0001)。
西伦等人(1998)报道了对来自欧洲和中东的 16 个 SLS 家族的研究,结果鉴定了 ALDH3A2 基因中的 11 个不同突变。在他们的研究中表征的突变谱包括 5 个导致氨基酸变化的核苷酸替换、5 个引入终止密码子的移码突变和 1 个在同一位置插入的框内缺失。还鉴定了多态性。突变广泛分布在整个基因中。
